

Evaluación del riesgo cardiovascular, y de sus determinantes, en pacientes con artritis reumatoide, espondilitis anquilosante y artritis psoriásica
INTRODUCCIÓN
La predicción del riesgo cardiovascular
Las enfermedades cardiovasculares (CV) siguen siendo una de las principales causas de morbilidad y mortalidad en Europa y Norteamérica. Los factores de riesgo CV clásico son la edad, el sexo, la hipertensión, el tabaquismo, el colesterol y la diabetes. Factores como la obesidad, la hipertrofia ventricular izquierda, historia familiar de cardiopatía coronaria prematura o la utilización de terapia hormonal sustitutiva también han sido considerados en la definición de riesgo CV. A partir de datos de estudios poblacionales se han creado tablas de predicción de la aparición de eventos CV a varios años. La estimación del riesgo CV en base a estas tablas tiene dos utilidades principales: identificar a los pacientes de alto riesgo en prevención primaria y ayudar en la toma de decisiones para la intervención farmacológica de la hipertensión arterial y la hipercolesterolemia.
En España, como en el resto de Europa, el cálculo del riesgo de presentar una enfermedad coronaria se ha basado en la función de Framingham [1, 2, 3], aunque se ha constatado que esta función sobrestima el riesgo en algunas de las poblaciones estudiadas, especialmente en países del entorno mediterráneo [4, 5]. De ahí el interés por encontrar una función que se adapte a la prevalencia real de factores de riesgo e incidencia de enfermedades CV de los diferentes países. En este contexto han ido apareciendo diversos sistemas para calcular el riesgo CV en nuestro país, entre los que se encuentran las tablas de riesgo coronario de Framingham calibradas (REGICOR)[6] y adaptadas (DORICA)[7] para la población española. También recientemente se han publicado las tablas del SCORE (Systematic Coronary Risk Evaluation)[8], que estiman el riesgo de muerte CV y son las actualmente recomendadas por las Sociedades Europeas y por el Comité Español Interdisciplinario para la Prevención Cardiovascular (CEIPC). No siendo la adaptación española de la tabla de Framingham (REGICOR) ni el SCORE perfectos, Buitrago et al demostraron que la tabla del SCORE se aproxima más al riesgo real en una cohorte española y presenta mejores criterios de validez [9]. Finalmente se ha calibrado la tabla del SCORE para población española [10]. Como puede observarse en la Figura 1, esta tabla se basa en el sexo, la edad, la tensión arterial sistólica, el tabaquismo y el colesterol total para predecir el riesgo de muerte por causa CV a 10 años.
La relación artritis reumatoide-inflamación vascular
Estudios recientes ponen de manifiesto una mayor incidencia de eventos CV, y de mortalidad de causa CV, en pacientes con artritis reumatoide (AR) frente a población de la misma edad y sexo [11, 12, 13]. Este aumento del riesgo de enfermedad CV es, con una gran probabilidad, consecuencia de un proceso de aterosclerosis acelerada [14, 15, 16].
La ateroesclerosis que acompaña a la AR ha sido demostrada en diferentes estadios evolutivos de la enfermedad [17, 18, 19]. La mayor incidencia de eventos CV sólo es parcialmente explicable por la presencia de factores clásicos de aterogénesis [13, 20, 21], jugando la inflamación un papel primordial en el desarrollo de este proceso. En este sentido, se ha demostrado una estrecha correlación entre los marcadores de inflamación y la presencia de aterosclerosis subclínica, valorada ésta por ultrasonografía de carótida [22, 23], y el desarrollo de eventos CV en el seguimiento de pacientes con AR de larga evolución [24].
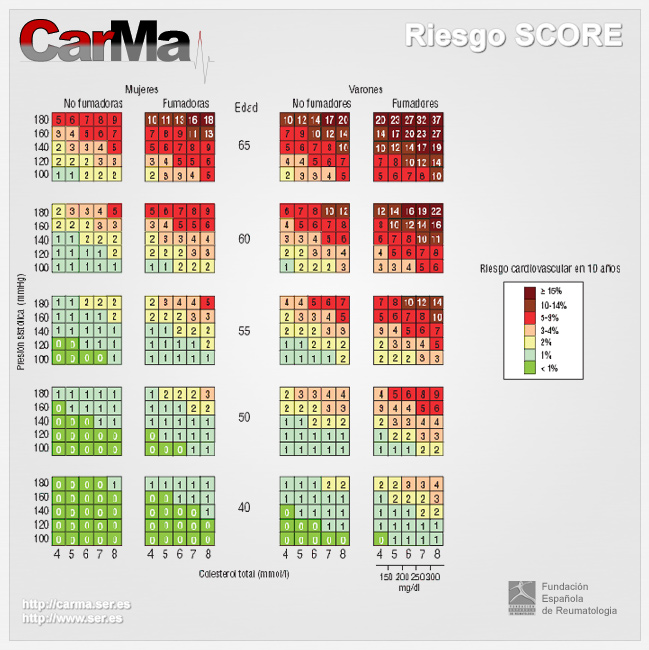
Figura 1. Tablas de riesgo SCORE calibradas para la población española [10]
Inflamación vascular en otras enfermedades inflamatorias reumáticas
De manera similar a lo descrito para pacientes con AR, datos recientes indican que los pacientes con espondiloartropatías, en concreto con artritis psoriásica (APs) y espondilitis anquilosante (EA), también pudieran presentar un aumento de riesgo CV [25]. Como en la AR, el aumento del riesgo CV en pacientes con espondiloartritis no puede ser totalmente explicado por la presencia de factores de riesgo clásicos de aterosclerosis [26]. Los resultados de un estudio publicado este año han mostrado que la población escandinava con espondiloartritis requiere cirugía coronaria a una edad más temprana que las personas sin esta enfermedad reumática [27]. Además, el riesgo de cirugía coronaria en pacientes con espondiloartritis es independiente de la presencia de factores de riesgo clásicos de aterosclerosis [27].
La APs es una enfermedad reumática inflamatoria crónica asociada a psoriasis, generalmente seronegativa para el factor reumatoide, y que se clasifica generalmente dentro del grupo de las espondiloartropatías. La psoriasis es una enfermedad inflamatoria crónica de la piel que habitualmente presenta una elevada prevalencia de factores clásicos de aterogénesis, especialmente en los casos con afectación cutánea grave [28]. De hecho, se ha comprobado que los pacientes con psoriasis cutánea aislada presentan un riesgo significativamente elevado de mortalidad CV en comparación con la población control [29, 30, 31]. Por otro lado, se ha observado que los pacientes con APs presentan un aumento de la aterosclerosis subclínica esperada que es independiente de la presencia de factores de riesgo CV clásicos [32, 33]. En este sentido, se ha descrito recientemente la existencia de diferentes estadios de enfermedad CV subclínica —disfunción endotelial y aterosclerosis carotídea subclínica— en población española con APs [32, 33].
La EA es una patología inflamatoria crónica que afecta a las articulaciones sacroiliacas y a las de la columna vertebral principalmente y que se observa en hasta el 1% de la población [34]. Los pacientes con EA tienen una mortalidad aproximadamente el doble de la encontrada en la población general, debido probablemente al elevado riesgo de desarrollo de eventos CV [26, 35, 36]. Aunque en la EA existen alteraciones cardiacas específicas como daño valvular aórtico o alteraciones en la conducción cardiaca [37, 38], la responsable del incremento de riesgo CV, al igual que en la AR o en la APs, pudiera ser una aterosclerosis acelerada [39, 40]. Esto es porque, como se ha descrito en la AR o en la APs, la aterosclerosis acelerada en pacientes con EA sólo puede ser explicada parcialmente por factores clásicos de aterogénesis como la dislipemia o la hipertensión arterial [39, 40]. Por otra parte, la presencia de una situación de inflamación crónica, característica de la EA, puede hacer a estos pacientes más propensos al desarrollo de aterosclerosis y eventos CV.
Parece lógico pensar que la AR, la APs y la EA por sí mismas también pueden ser un factor de riesgo CV independiente de los factores clásicos de aterosclerosis.
Determinantes genéticos e inflamatorios del riesgo cardiovascular
La AR es una enfermedad autoinmune donde los factores genéticos desempeñan un papel fundamental. Estudios de familias apoyan esta influencia genética, habiéndose descrito una mayor concordancia para el desarrollo de AR en gemelos univitelinos que en el resto de hermanos. En la AR se ha observado que varios genes dentro del complejo mayor de histocompatibilidad (MHC) influyen en la susceptibilidad para el desarrollo de AR en diferentes poblaciones. Una serie de alelos HLA-DRB1, dentro de la región de clase II del MHC, codifican una secuencia común de aminoácidos correspondientes a los residuos 67-74 de la cadena DRBeta1. Estos alelos, denominados “epítopo compartido”, se han asociado a una mayor susceptibilidad a desarrollar AR y también a una mayor gravedad en la expresión clínica de la AR, con más erosiones y manifestaciones extra-articulares.
Se ha observado una asociación entre el desarrollo de disfunción endotelial, considerada una fase precoz en el desarrollo de aterosclerosis, y la positividad de los alelos HLA-DRB1*04 “epítopo compartido” [17, 21]. Los pacientes con AR homocigóticos para el epítopo compartido tienen mayor riesgo de desarrollar una disfunción endotelial grave y los portadores del alelo HLA-DRB1*0404 tienen con mayor frecuencia disfunción del endotelio vascular que el resto de los pacientes con AR [21].
Y no sólo se asocia a disfunción endotelial, sino que también se observa una incidencia aumentada de eventos CV en pacientes HLA-DRB1*04 positivos para el epítopo compartido, más clara si cabe en asociación al alelo DRB1*0404 [21]. En el Reino Unido se ha descrito además un aumento de la mortalidad de causa CV por cardiopatía isquémica en AR portadoras del epítopo compartido, en particular en relación con los genotipos HLA-DRB1*0101/*0401 o 0404/*0404 [41, 42].
Aunque los resultados descritos arriba enfatizan la importancia de la región HLA en el desarrollo de aterogénesis acelerada en los pacientes con AR, es importante tener en cuenta que la AR es una enfermada poligénica y que la contribución de genes dentro del MHC para la susceptibilidad y desarrollo de AR solo es de alrededor de 1/3 a 1/4 de la contribución genética total [43]. Debido a ello, para un mayor conocimiento de las bases genéticas implicadas en el proceso aterogénico de la AR es importante que se determinen otros genes potencialmente candidatos que puedan influir en el proceso aterogénico en estos pacientes.
Numerosos estudios han señalado al gen PTPN22 como un factor genético de susceptibilidad a diferentes enfermedades autoinmunes, incluida la AR. De hecho, el gen PTPN22 es la asociación genética con la AR mejor establecida y replicada, aparte de los genes HLA [43]. De forma muy interesante un trabajo reciente ha reportado que el gen PTPN22, además de predisponer a autoinmunidad, potencia el desarrollo de aterosclerosis [44] y de esta forma une estos dos fenómenos, por lo que sería de gran interés estudiar el gen PTPN22, junto con los genes HLA-DR, en los procesos CV de la AR.
En esta línea se ha sugerido que los procesos inmunológicos e inflamatorios podrían ser importantes en el desarrollo o desestabilización de la placa de aterogénesis [45]. Uno de estos mecanismos que ha despertado gran interés es la señalización vía receptores tipo toll (toll like receptors - TLR) y su influencia en el desarrollo de aterogénesis [45]. Los TLRs son una familia de receptores (se han identificado hasta 12 receptores en mamíferos) conocidos como “receptores de reconocimiento de patrón” y cuya función principal es el inicio de una respuesta inmune innata eficaz ante patógenos y otros estímulos. Este tipo de receptores, en concreto TLR4, TLR1, y TLR2 se expresan en modelos de ratón de aterosclerosis así como en placas aterogénicas de humanos [45]. Datos muy recientes destacan la implicación del TLR4 en la iniciación y progresión de aterosclerosis [46]. El TLR4 se expresa en distintos tipos celulares de la pared de vasos sanguíneos ateroscleróticos como células endoteliales, macrófagos, fibroblastos y células dendríticas [47]. El TLR4 presenta dos polimorfismos que afectan a su dominio extracelular y que generan un cambio aminoacídico: Asp299Gly y Thr399Ile. Ambos polimorfismos se han relacionado con una menor respuesta ante estímulos bacterianos [48] y por lo tanto con una menor inflamación a nivel sistémico, lo cual podría constituir un efecto protector para el riesgo CV. De hecho, se ha observado que el polimorfismo Asp299Gly se asocia con un menor riesgo de padecer aterosclerosis de la carótida así como con un menor grosor de la capa íntima-media de esta arteria [49].
Además se sabe que durante la formación y progresión de la placa de aterogénesis intervienen mecanismos proinflamatorios. La exposición de la superficie endotelial a distintos factores de riesgo, como infecciones o inflamación crónica, resulta en daño y en inflamación local del tejido endotelial. Se produce una interacción entre células T y macrófagos con secreción de citoquinas proinflamatorias, como el factor de necrosis tumoral alpha (TNFa) o la interleukina 1 (IL-1), resultando en la transformación del endotelio hacia un estado protrombótico, caracterizado por un aumento en la expresión de moléculas de adhesión y de quimioquinas. Muchas de estas moléculas se consideran biomarcadores indicativos de una enfermedad aterosclerótica activa, es el caso de la proteína C reactiva (CRP), la cual estimula la secreción de moléculas de adhesión y quimioquinas, o de la IL-6, ICAM-1, TNFa, y VCAM-1 [50]. En un estudio reciente se han observado niveles elevados de estos biomarcadores en pacientes con AR en comparación con población sana y una asociación entre los niveles de VCAM-1 y el espesor de la íntima media carotídea [50]. Algunos polimorfismos en genes pueden afectar los niveles de expresión de estas moléculas y se han asociado a enfermedades CV, como es el caso de polimorfismos CRP 1059 G/C, IL-6– 174G/C, MIF– 173G/C, ICAM-1– 469E/K, y E-sel Ser128Arg, que se han asociado con un mayor riesgo de padecer aterosclerosis o angina [51, 52]. Recientemente se ha informado de la existencia de variantes polimórficas del gen CRP que alteran la actividad transcripcional del mismo y que se encuentran relacionados con los niveles basales en suero de la proteína C reactiva [53, 54].
El óxido nítrico (ON) es un importante mediador en la aparición de la disfunción endotelial previa al daño vascular. En los vasos, el ON es producido por la expresión constitutiva de la enzima óxido nítrico sintasa endotelial (eNOS) o bien tras la activación por diversos estímulos de la óxido nítrico sintasa inducible (iNOS o NOS2). El ON tiene multitud de funciones en el organismo, pero su capacidad de ejercer cambios funcionales y morfológicos sobre la pared de los vasos sanguíneos, como la vasodilatación dependiente de endotelio (VED), es esencial en el mantenimiento de la homeostasis vascular y en la prevención de procesos de aterogénesis. La producción de ON por la eNOS permite un flujo sanguíneo adecuado inhibiendo la contracción de la musculatura lisa de los vasos sanguíneos y así también inhibe la generación de la placa de ateroma. En los vasos aterogénicos la vasodilatación dependiente de endotelio está alterada y esto se cree es debido a una inhibición de la actividad de la eNOS [55]. El papel de la iNOS en la aterogénesis no está claramente definido, pero se ha observado un aumento de los niveles de ARN mensajero de esta enzima en macrófagos y células del músculo liso de los vasos sanguíneos aterogénicos. Se especula que el exceso en la producción de NO que mediaría la iNOS podría aumentar el daño celular y así contribuir a la muerte celular y necrosis de tejidos que aparece en las lesiones ateroscleróticas avanzadas [55]. Los niveles de producción de estas enzimas se pueden ver afectados por polimorfismos genéticos en sus regiones reguladoras. En el exón 7 del gen de la eNOS se ha descrito un polimorfismo que genera un cambio aminoacídico de ácido glutámico por ácido aspártico (-786T/C ó Glu298Asp) que se ha asociado con predisposición a distintas enfermedades cardiovasculares y que se cree que afecta a la actividad de la enzima [56, 57]. En la región promotora del gen de la iNOS se han descrito numerosos polimorfismos, entre ellos destaca un microsatélite bialélico TAAA que afecta a la actividad del promotor de la iNOS y que además se ha asociado con enfermedades cardiovasculares [58].
Podemos concluir que el estudio a nivel genético y molecular pone de manifiesto una estrecha relación entre moléculas inflamatorias relacionadas etiopatogénicamente con las enfermedades reumáticas y con las enfermedades CV.
Justificación del estudio
Un estudio que permita establecer el riesgo de muerte por causa CV en población con enfermedades reumáticas inflamatorias y que permita adaptar o calibrar las tablas de riesgo en uso sería de gran utilidad, puesto que habitualmente se toman decisiones de iniciar un tratamiento hipolipemiante o antihipertensivo en pacientes con enfermedades reumáticas inflamatorias basándonos en riesgo de población sin el riesgo de la inflamación crónica.
- ¿Es necesario bajar los niveles de colesterol, o de tensión arterial o la edad a la que se debería empezar a tratar?
- ¿Hay otros factores que se deban añadir a las tablas de riesgo y que mejoren la utilidad predictiva en nuestros pacientes?
- ¿Es igual el riesgo en todas las artritis?
- ¿Hasta qué punto los factores genéticos que se proponen están presentes en población con o sin enfermedades inflamatorias articulares o sistémicas entendiendo por sistémicas a la inflamación vascular?
Desarrollo |
Patrocinio |
 |  |